石墨烯的结构
首先,我们需要在openmx中模拟出石墨烯的结构。详见之前的博文
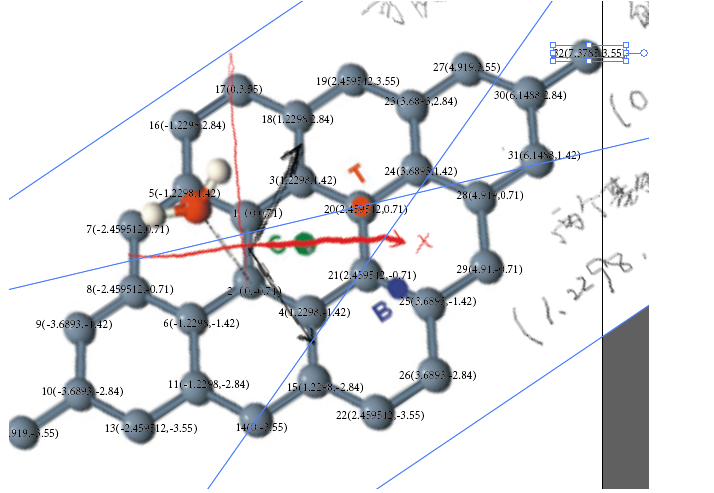
用作气体吸附的模型,我们不仅仅需要原胞,更需要更加灵活的晶胞设置。引用上文中的一幅插图
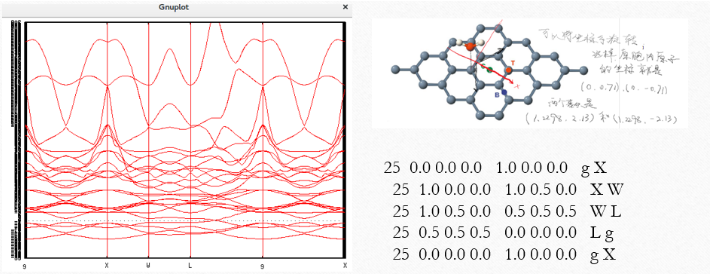
我们在本文中使用上面图片中石墨烯的原胞,表面上有32个原子,实际上经过我的核算有20个原子的原胞。
原本的原胞有如下
Atoms.Number 2
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C 0.000000000 0.710000000 0.000000000 2.0 2.0
2 C 0.000000000 -0.71000000 0.000000000 2.0 2.0
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors # unit=Ang.
1.22980000000000 2.13000000000000 0.00000000000000
1.22980000000000 -2.13000000000000 0.00000000000000
0.00000000000000 0.00000000000000 20.00000000000000
Atoms.UnitVectors>原子位于原点两侧。
把原胞扩展到32个的Graphene_8.dat,
#
# File Name
#
System.CurrrentDirectory ./ # default=./
System.Name Graphene_8
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 1 # default=1 (0-2)
#
# Definition of Atomic Species
#
Species.Number 1
<Definition.of.Atomic.Species
C C6.0-s2p2d1 C_PBE13
Definition.of.Atomic.Species>
#
# Atoms
#
Atoms.Number 32
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C 0.00000000 0.710000000 0.000000000 2.0 2.0
2 C 0.00000000 -0.71000000 0.000000000 2.0 2.0
3 C 1.22980000 1.420000000 0.000000000 2.0 2.0
4 C 1.22980000 -1.42000000 0.000000000 2.0 2.0
5 C -1.2298000 1.420000000 0.000000000 2.0 2.0
6 C -1.2298000 -1.42000000 0.000000000 2.0 2.0
7 C -2.4559120 0.71000000 0.000000000 2.0 2.0
8 C -2.4559120 -0.71000000 0.000000000 2.0 2.0
9 C -3.6893000 -1.42000000 0.000000000 2.0 2.0
10 C -3.6893000 -2.84000000 0.000000000 2.0 2.0
11 C -1.2298000 -2.84000000 0.000000000 2.0 2.0
12 C -4.9190000 -3.55000000 0.000000000 2.0 2.0
13 C -2.4595120 -3.55000000 0.000000000 2.0 2.0
14 C 0.00000000 -3.55000000 0.000000000 2.0 2.0
15 C 1.22980000 -2.84000000 0.000000000 2.0 2.0
16 C -1.2298000 2.840000000 0.000000000 2.0 2.0
17 C 0.00000000 3.550000000 0.000000000 2.0 2.0
18 C 1.22980000 2.840000000 0.000000000 2.0 2.0
19 C 2.45951200 3.550000000 0.000000000 2.0 2.0
20 C 2.45951200 0.710000000 0.000000000 2.0 2.0
21 C 2.45951200 -0.71000000 0.000000000 2.0 2.0
22 C 2.45951200 -3.55000000 0.000000000 2.0 2.0
23 C 3.68930000 2.840000000 0.000000000 2.0 2.0
24 C 3.68930000 1.420000000 0.000000000 2.0 2.0
25 C 3.68930000 -1.42000000 0.000000000 2.0 2.0
26 C 3.68930000 -2.84000000 0.000000000 2.0 2.0
27 C 4.91900000 3.550000000 0.000000000 2.0 2.0
28 C 4.91900000 0.710000000 0.000000000 2.0 2.0
29 C 4.91900000 -0.71000000 0.000000000 2.0 2.0
30 C 6.14880000 2.840000000 0.000000000 2.0 2.0
31 C 6.14880000 1.420000000 0.000000000 2.0 2.0
32 C 7.37850000 3.550000000 0.000000000 2.0 2.0
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors # unit=Ang.
9.83800000000000 0000000000000000 0.00000000000000
4.91900000000000 8.52000000000000 0.00000000000000
0.00000000000000 0.00000000000000 100.00000000000000
Atoms.UnitVectors>
#
# SCF or Electronic System
#
scf.XcType GGA-PBE # LDA|LSDA-CA|LSDA-PW|GGA-PBE
scf.SpinPolarization Off # On|Off|NC
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 200.0 # default=150 (Ry)
scf.maxIter 60 # default=40
scf.EigenvalueSolver band # DC|GDC|Cluster|Band
scf.Kgrid 5 5 1 # means 4x4x4
scf.Mixing.Type rmm-diis # Simple|Rmm-Diis|Gr-Pulay|Kerker|Rmm-Diisk
scf.Init.Mixing.Weight 0.010 # default=0.30
scf.Min.Mixing.Weight 0.001 # default=0.001
scf.Max.Mixing.Weight 0.002 # default=0.40
scf.Mixing.History 7 # default=5
scf.Mixing.StartPulay 5 # default=6
scf.criterion 1.0e-9 # default=1.0e-6 (Hartree)
#
# MD or Geometry Optimization
#
MD.Type nomd # Nomd|Opt|NVE|NVT_VS|NVT_NH
MD.maxIter 1 # default=1
MD.TimeStep 0.5 # default=0.5 (fs)
MD.Opt.criterion 1.0e-4 # default=1.0e-4 (Hartree/bohr)
Band.dispersion on
Band.Nkpath 3
<Band.kpath
25 0.5 0.0 0.0 0.333 0.333 0.0 M K
25 0.333 0.333 0.0 0.0 0.0 0.0 K G
25 0.0 0.0 0.0 0.5 0.0 0.0 G M
Band.kpath>
#
# partial charge calculation
#
partial.charge on # on|off, default=off
partial.charge.energy.window 2.0 # in eV
#
# DOS and PDOS
#
Dos.fileout on # on|off, default=off
Dos.Erange -20.0 20.0 # default = -20 20
Dos.Kgrid 10 10 1 # default = Kgrid1 Kgrid2 Kgrid3它所对应的坐标图片在下面:
可是这样做出来的能带图过于杂乱。
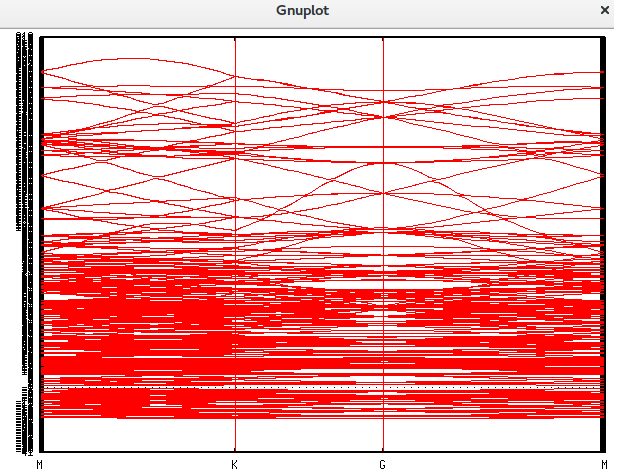
正确的晶胞选择
奈何感觉这个晶胞选取并正确,因为我们仔细放大上图中的费米能级附近的能带,发现K点附近并没有达到零带隙。
应该重新分析那个原胞的选取,第一,通过我的计算,原胞包含18个原子,分别包括中心的8个,角上的8/4=2个,和边上的16/2=8个,加起来一共是18个。
想办法在上面的晶胞中选取18个原子,最简单的方法就是去掉上边和右边的一排。
经过了几次迭代,得到Graphene_11.dat如下:
#
# File Name
#
System.CurrrentDirectory ./ # default=./
System.Name Graphene_11
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 1 # default=1 (0-2)
#
# Definition of Atomic Species
#
Species.Number 1
<Definition.of.Atomic.Species
C C6.0-s2p2d1 C_PBE13
Definition.of.Atomic.Species>
#
# Atoms
#
Atoms.Number 18
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C 0.00000000 0.710000000 0.000000000 2.0 2.0
2 C 0.00000000 -0.71000000 0.000000000 2.0 2.0
3 C 1.22980000 1.420000000 0.000000000 2.0 2.0
4 C 1.22980000 -1.42000000 0.000000000 2.0 2.0
5 C -1.2298000 1.420000000 0.000000000 2.0 2.0
6 C -1.2298000 -1.42000000 0.000000000 2.0 2.0
7 C -2.4559120 0.71000000 0.000000000 2.0 2.0
8 C -2.4559120 -0.71000000 0.000000000 2.0 2.0
9 C -3.6893000 -1.42000000 0.000000000 2.0 2.0
10 C -3.6893000 -2.84000000 0.000000000 2.0 2.0
11 C -1.2298000 -2.84000000 0.000000000 2.0 2.0
12 C -4.9190000 -3.55000000 0.000000000 2.0 2.0
13 C -2.4595120 -3.55000000 0.000000000 2.0 2.0
14 C 0.00000000 -3.55000000 0.000000000 2.0 2.0
15 C 1.22980000 -2.84000000 0.000000000 2.0 2.0
16 C 2.45951200 0.710000000 0.000000000 2.0 2.0
17 C 2.45951200 -0.71000000 0.000000000 2.0 2.0
18 C 3.68930000 1.420000000 0.000000000 2.0 2.0
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors # unit=Ang.
4.91900000000000 -8.5200000000000 0.00000000000000
4.91900000000000 8.52000000000000 0.00000000000000
0.00000000000000 0.00000000000000 100.000000000000
Atoms.UnitVectors>
#
# SCF or Electronic System
#
scf.XcType GGA-PBE # LDA|LSDA-CA|LSDA-PW|GGA-PBE
scf.SpinPolarization Off # On|Off|NC
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 200.0 # default=150 (Ry)
scf.maxIter 60 # default=40
scf.EigenvalueSolver band # DC|GDC|Cluster|Band
scf.Kgrid 5 5 1 # means 4x4x4
scf.Mixing.Type rmm-diis # Simple|Rmm-Diis|Gr-Pulay|Kerker|Rmm-Diisk
scf.Init.Mixing.Weight 0.010 # default=0.30
scf.Min.Mixing.Weight 0.001 # default=0.001
scf.Max.Mixing.Weight 0.002 # default=0.40
scf.Mixing.History 7 # default=5
scf.Mixing.StartPulay 5 # default=6
scf.criterion 1.0e-9 # default=1.0e-6 (Hartree)
#
# MD or Geometry Optimization
#
MD.Type nomd # Nomd|Opt|NVE|NVT_VS|NVT_NH
MD.maxIter 1 # default=1
MD.TimeStep 0.5 # default=0.5 (fs)
MD.Opt.criterion 1.0e-4 # default=1.0e-4 (Hartree/bohr)
Band.dispersion on
Band.Nkpath 3
<Band.kpath
25 0.5 0.0 0.0 0.333 0.333 0.0 M K
25 0.333 0.333 0.0 0.0 0.0 0.0 K G
25 0.0 0.0 0.0 0.5 0.0 0.0 G M
Band.kpath>
#
# partial charge calculation
#
partial.charge on # on|off, default=off
partial.charge.energy.window 2.0 # in eV
#
# DOS and PDOS
#
Dos.fileout on # on|off, default=off
Dos.Erange -20.0 20.0 # default = -20 20
Dos.Kgrid 10 10 1 # default = Kgrid1 Kgrid2 Kgrid3以及它所对应的能带图
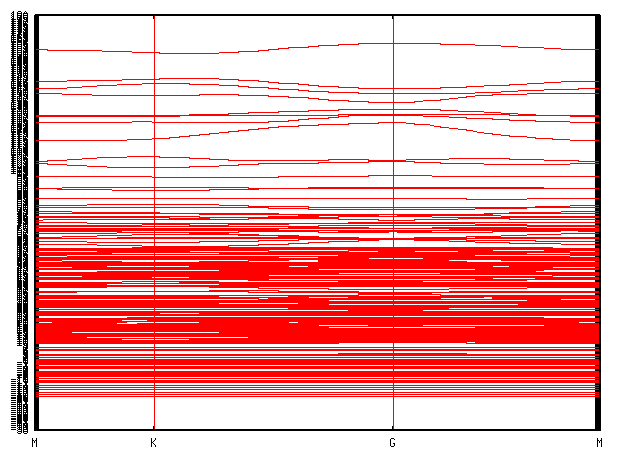
通过放大的图可以看到能带和原胞能带符合得不错,只是变得密了。在K点也有零带隙的表现,所以这个就可以认为是正确的了。
研究步骤
在上面的准备工作中已经完成了石墨烯晶胞的构建,下面需要将掺杂引入。第一个问题就是选用什么样的PAO贋原子轨道和VPS贋势。
从上面的Graphene_11.dat中可以看到,C原子选取的是C C6.0-s2p2d1 C_PBE13,这里的6.0指的是截断半径,我们打算仿真磷在石墨烯表面的掺杂,所以需要寻找P原子的信息。在openmx3.8的源码目录下找到DFT_DATA13文件夹,下面有PAO和VPS库。看到PAO库里有C6.0.pao文件,这就是C6.0-s2p2d1的基础。
找到P8.0.pao文件,里面有P原子的基本描述:
AtomSpecies 15
max.occupied.N 3
total.electron 15.0
valence.electron 5.0
<occupied.electrons
1 2.0
2 2.0 6.0
3 2.0 3.0 0.0
occupied.electrons>对于磷原子我们暂时选取P8.0-s2p4d1,贋势选择P_CA13。通过修改Graphene_11.dat得到Graphene_12.dat,增加了一个原点上方的磷原子,我们发现在费米能级附近的能带更加密集、更加平直。
研究吸附主要是研究几个问题:
1.最优的几何结构,怎么吸附最稳定
建立bash脚本,让磷原子在石墨烯表面不同高度进行弛豫,以判断最好的位置。
#!bin/bash
if [ ! -f "energy.dat" ];then
echo 'energy.dat not exist'
else
rm -rf energy.dat
fi
for d in 0.5 0.6 0.7 0.8 0.9 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 2.0
do
cat << EOF > Graphene_13.dat
#
# File Name
#
System.CurrrentDirectory ./ # default=./
System.Name Graphene_13
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 1 # default=1 (0-2)
#
# Definition of Atomic Species
#
Species.Number 2
<Definition.of.Atomic.Species
C C6.0-s2p2d1 C_PBE13
P P8.0-s2p2d1 P_CA13
Definition.of.Atomic.Species>
#
# Atoms
#
Atoms.Number 19
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C 0.00000000 0.710000000 0.000000000 2.0 2.0
2 C 0.00000000 -0.71000000 0.000000000 2.0 2.0
3 C 1.22980000 1.420000000 0.000000000 2.0 2.0
4 C 1.22980000 -1.42000000 0.000000000 2.0 2.0
5 C -1.2298000 1.420000000 0.000000000 2.0 2.0
6 C -1.2298000 -1.42000000 0.000000000 2.0 2.0
7 C -2.4559120 0.71000000 0.000000000 2.0 2.0
8 C -2.4559120 -0.71000000 0.000000000 2.0 2.0
9 C -3.6893000 -1.42000000 0.000000000 2.0 2.0
10 C -3.6893000 -2.84000000 0.000000000 2.0 2.0
11 C -1.2298000 -2.84000000 0.000000000 2.0 2.0
12 C -4.9190000 -3.55000000 0.000000000 2.0 2.0
13 C -2.4595120 -3.55000000 0.000000000 2.0 2.0
14 C 0.00000000 -3.55000000 0.000000000 2.0 2.0
15 C 1.22980000 -2.84000000 0.000000000 2.0 2.0
16 C 2.45951200 0.710000000 0.000000000 2.0 2.0
17 C 2.45951200 -0.71000000 0.000000000 2.0 2.0
18 C 3.68930000 1.420000000 0.000000000 2.0 2.0
19 P 0.00000000 0.000000000 $d 2.5 2.5
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors # unit=Ang.
4.91900000000000 -8.5200000000000 0.00000000000000
4.91900000000000 8.52000000000000 0.00000000000000
0.00000000000000 0.00000000000000 100.000000000000
Atoms.UnitVectors>
#
# SCF or Electronic System
#
scf.XcType GGA-PBE # LDA|LSDA-CA|LSDA-PW|GGA-PBE
scf.SpinPolarization Off # On|Off|NC
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 200.0 # default=150 (Ry)
scf.maxIter 60 # default=40
scf.EigenvalueSolver band # DC|GDC|Cluster|Band
scf.Kgrid 5 5 1 # means 4x4x4
scf.Mixing.Type rmm-diis # Simple|Rmm-Diis|Gr-Pulay|Kerker|Rmm-Diisk
scf.Init.Mixing.Weight 0.010 # default=0.30
scf.Min.Mixing.Weight 0.001 # default=0.001
scf.Max.Mixing.Weight 0.002 # default=0.40
scf.Mixing.History 7 # default=5
scf.Mixing.StartPulay 5 # default=6
scf.criterion 1.0e-9 # default=1.0e-6 (Hartree)
#
# MD or Geometry Optimization
#
MD.Type nomd # Nomd|Opt|NVE|NVT_VS|NVT_NH
MD.maxIter 1 # default=1
MD.TimeStep 0.5 # default=0.5 (fs)
MD.Opt.criterion 1.0e-4 # default=1.0e-4 (Hartree/bohr)
Band.dispersion on
Band.Nkpath 3
<Band.kpath
25 0.5 0.0 0.0 0.333 0.333 0.0 M K
25 0.333 0.333 0.0 0.0 0.0 0.0 K G
25 0.0 0.0 0.0 0.5 0.0 0.0 G M
Band.kpath>
#
# partial charge calculation
#
partial.charge on # on|off, default=off
partial.charge.energy.window 2.0 # in eV
#
# DOS and PDOS
#
Dos.fileout on # on|off, default=off
Dos.Erange -20.0 20.0 # default = -20 20
Dos.Kgrid 10 10 1 # default = Kgrid1 Kgrid2 Kgrid3
EOF
#if [ ! -f 'Graphene_13.out' ];then
./openmx Graphene_13.dat
#fi
echo "d=$d " >> energy.dat
E=$(grep "Utot" Graphene_13.out|head -1| awk '{print $2}')
echo "E=$E " >> energy.dat
done这里把距离和能量输出到一个文件中,再进行人为判断。经过计算我们把计算结果粘贴出来:
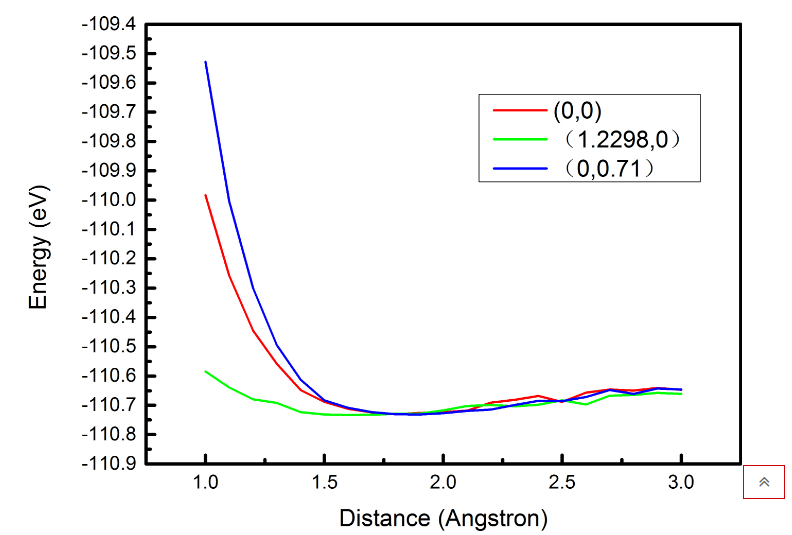
这里要强调一点,磷原子的XoY坐标为(0,0)
| Distance | Energy |
| 埃 | eV |
| 0.5 | -106.4346033 |
| 0.6 | -107.4488502 |
| 0.7 | -108.3255414 |
| 0.8 | -109.0490693 |
| 0.9 | -109.5908616 |
| 1 | -109.9831407 |
| 1.1 | -110.2575064 |
| 1.2 | -110.4447311 |
| 1.3 | -110.5581597 |
| 1.4 | -110.6475163 |
| 1.5 | -110.6882315 |
| 1.6 | -110.7118016 |
| 1.7 | -110.7245302 |
| 1.8 | -110.7300852 |
| 1.9 | -110.7258409 |
| 2 | -110.7232356 |
| 2.1 | -110.7179988 |
| 2.2 | -110.6904221 |
| 2.3 | -110.6811691 |
| 2.4 | -110.66832 |
| 2.5 | -110.6884847 |
| 2.6 | -110.6566485 |
| 2.7 | -110.6458464 |
| 2.8 | -110.6499256 |
| 2.9 | -110.6405059 |
| 3 | -110.6472704 |
换一个点磷原子(1.2298,0)
| 0.5 | -110.1456904 |
| 0.6 | -110.2453609 |
| 0.7 | -110.3366086 |
| 0.8 | -110.412732 |
| 0.9 | -110.5144254 |
| 1 | -110.5844483 |
| 1.1 | -110.6391902 |
| 1.2 | -110.6792789 |
| 1.3 | -110.6916974 |
| 1.4 | -110.7228873 |
| 1.5 | -110.7310903 |
| 1.6 | -110.7325368 |
| 1.7 | -110.7320324 |
| 1.8 | -110.7283038 |
| 1.9 | -110.7282692 |
| 2 | -110.7169384 |
| 2.1 | -110.701813 |
| 2.2 | -110.6984034 |
| 2.3 | -110.7031432 |
| 2.4 | -110.6975467 |
| 2.5 | -110.6827083 |
| 2.6 | -110.6973192 |
| 2.7 | -110.6668035 |
| 2.8 | -110.6644891 |
| 2.9 | -110.6570942 |
| 3 | -110.6611948 |
再换一个点磷原子(0,0.7)
| 0.5 | -101.7200994 |
| 0.6 | -104.5135856 |
| 0.7 | -106.3820138 |
| 0.8 | -107.7749174 |
| 0.9 | -108.8062766 |
| 1 | -109.5286926 |
| 1.1 | -110.0033858 |
| 1.2 | -110.2999951 |
| 1.3 | -110.4940053 |
| 1.4 | -110.6125845 |
| 1.5 | -110.6830201 |
| 1.6 | -110.7083819 |
| 1.7 | -110.7232931 |
| 1.8 | -110.7299741 |
| 1.9 | -110.7315692 |
| 2 | -110.7271532 |
| 2.1 | -110.7187878 |
| 2.2 | -110.7147709 |
| 2.3 | -110.6983154 |
| 2.4 | -110.6850805 |
| 2.5 | -110.685528 |
| 2.6 | -110.671795 |
| 2.7 | -110.6479944 |
| 2.8 | -110.6611288 |
| 2.9 | -110.6429204 |
| 3 | -110.6463351 |
在下面画出一个图来直观地展示出总能量随距离的变化
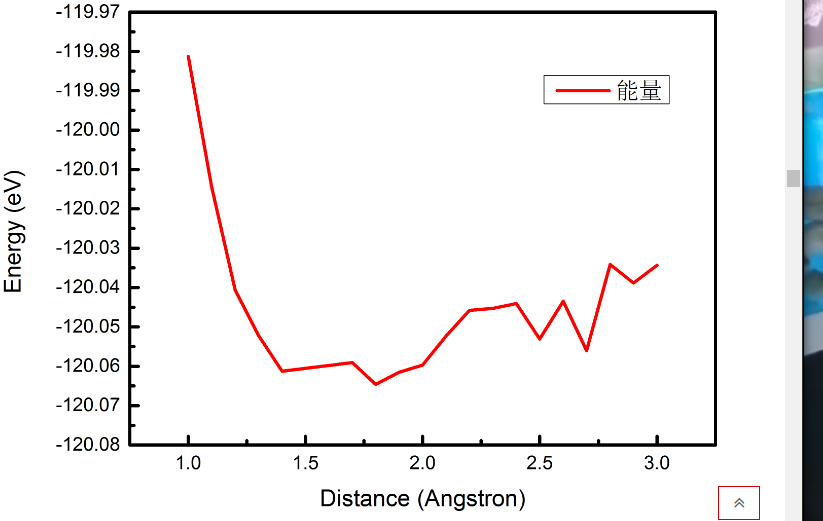
1.1氧原子的吸附
我们用上面的方法将磷原子换成氧原子,在(1.2298,0)这个点上进行仿真,得到了如下的能量-距离关系。
1.0 -119.981285544460
1.1 -120.014350570612
1.2 -120.040624352151
1.3 -120.052160956069
1.4 -120.061235898467
1.5 -120.073054788872
1.6 -120.069042528046
1.7 -120.059083301341
1.8 -120.064618046068
1.9 -120.061497831969
2.0 -120.059686309498
2.1 -120.052269602504
2.2 -120.045770598804
2.3 -120.045255715742
2.4 -120.044031918423
2.5 -120.053092740570
2.6 -120.043500583778
2.7 -120.056007311243
2.8 -120.034129674387
2.9 -120.038836825107
3.0 -120.034314288385
2.吸附能量变化
为了研究能量变化,我们设计了由C原子构成的石墨烯晶胞Graphene_11.dat,石墨烯上的磷原子组合的Graphene_12.dat,还有单独的文件Patom.dat。
我们在它们的.out文件中查找总能量Utot。
EGraphene_11 -103.940151228761
EGraphene_12 -110.647516282634
EPatom -6.726714934724
能量差也就是吸附过程放出能量 :
EGraphene_12 – (EGraphene_11 + EPatom ) = 0.0193 eV
3.电荷转移
对于态密度的画图,我们以Graphene_12项目来做例子,原子19是一个石墨烯面上吸附的磷原子。可以计算PDOS,并且用gnuplot画出来:
plot 'Graphene_12.PDOS.Tetrahedron.atom19' w l,'Graphene_12.PDOS.Tetrahedron.atom19.s1' w l,'Graphene_12.PDOS.Tetrahedron.atom19.p1' w l,'Graphene_12.PDOS.Tetrahedron.atom19.p2' w l,'Graphene_12.PDOS.Tetrahedron.atom19.p3' w l,'Graphene_12.PDOS.Tetrahedron.atom19.d1' w l如果用脚本来完成这个任务,可以用如下脚本:
set style data lines
set border lw 3
set zeroaxis lw 3
set ytics 5
set mytics 10
set term eps size 4,3
set output "Graphene_12.PDOS.Tetrahedron.atom19.eps
plot 'Graphene_12.PDOS.Tetrahedron.atom19' w l,\
'Graphene_12.PDOS.Tetrahedron.atom19.s1' w l,\
'Graphene_12.PDOS.Tetrahedron.atom19.p1' w l,\
'Graphene_12.PDOS.Tetrahedron.atom19.p2' w l,\
'Graphene_12.PDOS.Tetrahedron.atom19.p3' w l,\
'Graphene_12.PDOS.Tetrahedron.atom19.d1' w l
set output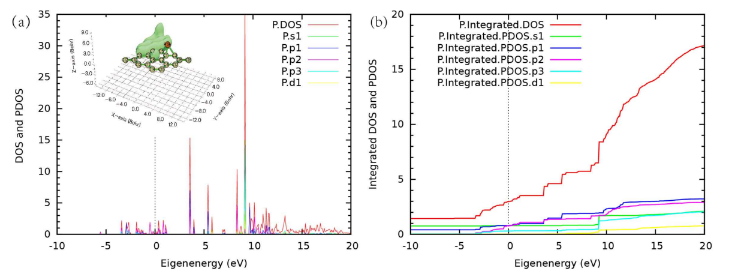
上面这个图是把磷原子放在0.00000000 1.22980000 1.520000000 这个位置也就是苯环中心的位置来计算出来的分波态密度。
在openmx中,输出的.cube文件可以使用multiwfn软件来查看,也可以用Gaussian软件来查看,关键是从linux虚拟机导出到windows时要进行一下压缩,否则会出现文件错误的问题。下面这张图就是Graphene_12.tden.cube在multiwfn中的展示效果。multiwfn虽然是开源的免费软件,但是在文章中使用的话,一定要引用
Tian Lu, Feiwu Chen, J. Comput. Chem., 33, 580-592 (2012).
这篇文章,否则会被加入黑名单。
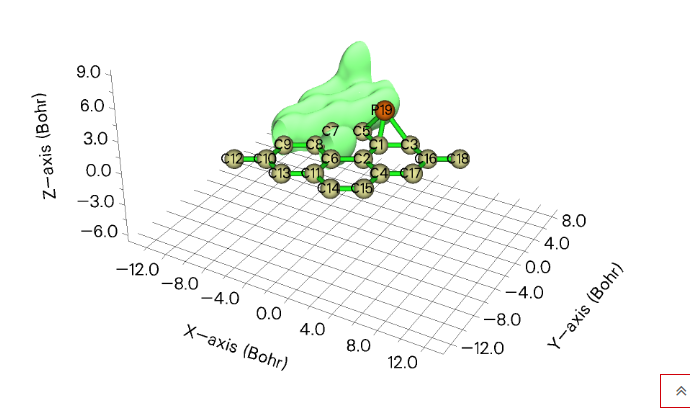
另外还可以把.cube文件用VMD(Visual Molecular Dynamics)打开,按照下面这篇文章的方法来:
在VMD里将cube文件瞬间绘制成效果极佳的等值面图的方法
画等值面。需要先去VMD官方网站下载安装,注册一下,直接在下载界面输入用户名和密码自动跳转进入注册界面。安装好后按照上面的文章来复制好脚本,修改VMD的根目录下的vmd.rc。最后打开VMD,在命令行中输入命令,即可画出等值面的图。这里还需要提一个事情,读进来的cube文件要把后缀改成cub。另外,对于不同原子选取不同颜色的问题,需要在VMD菜单栏选择Graphics选择colors,对不同原子设置不同颜色。
目前看来下面的一段自己编写的计算电荷转移的方法是错误的,得出了与multiwfn相反的结论。
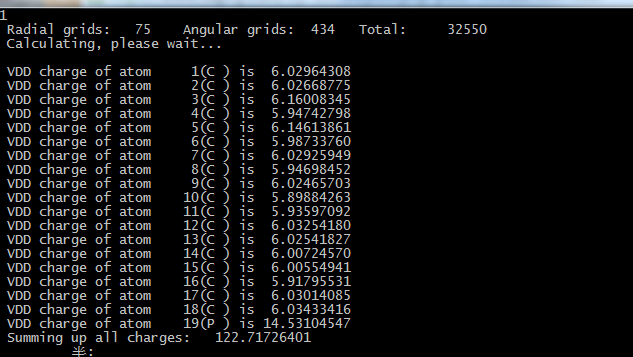
对于原子间电子的转移,可以参考.out文件,还是以Graphene_12为例,在.out文件中可以看到atom19的磷原子的电荷
19 P Up spin Down spin Sum Diff
multiple
s 0 0.928371279 0.928371279 1.856742559 0.000000000
sum over m 0.928371279 0.928371279 1.856742559 0.000000000
s 1 0.015926106 0.015926106 0.031852212 0.000000000
sum over m 0.015926106 0.015926106 0.031852212 0.000000000
sum over m+mul 0.944297385 0.944297385 1.888594771 0.000000000
px 0 0.523208816 0.523208816 1.046417632 0.000000000
py 0 0.422925608 0.422925608 0.845851217 0.000000000
pz 0 0.522705967 0.522705967 1.045411933 0.000000000
sum over m 1.468840391 1.468840391 2.937680782 0.000000000
px 1 0.014947075 0.014947075 0.029894151 0.000000000
py 1 0.023867769 0.023867769 0.047735538 0.000000000
pz 1 0.017900000 0.017900000 0.035800000 0.000000000
sum over m 0.056714844 0.056714844 0.113429689 0.000000000
sum over m+mul 1.525555236 1.525555236 3.051110471 0.000000000
d3z^2-r^2 0 0.045672672 0.045672672 0.091345344 0.000000000
dx^2-y^2 0 0.022454960 0.022454960 0.044909919 0.000000000
dxy 0 0.043444469 0.043444469 0.086888938 0.000000000
dxz 0 0.035721802 0.035721802 0.071443605 0.000000000
dyz 0 0.058941319 0.058941319 0.117882638 0.000000000
sum over m 0.206235222 0.206235222 0.412470444 0.000000000
sum over m+mul 0.206235222 0.206235222 0.412470444 0.000000000这里的multiple代表角向和径向的电子数投影。我们看s层的电子数直接找s下面的sum over m+mul就可以了,原子最外层核外电子可以用s、p和d层的sum over m + mul这一项的和。
我们对比一下孤立磷原子的电荷,就可以非常非常确定的看到电荷转移的存在。
1 P Up spin Down spin Sum Diff
multiple
s 0 0.999974266 0.999974266 1.999948532 0.000000000
sum over m 0.999974266 0.999974266 1.999948532 0.000000000
s 1 0.000025734 0.000025734 0.000051468 0.000000000
sum over m 0.000025734 0.000025734 0.000051468 0.000000000
sum over m+mul 1.000000000 1.000000000 2.000000000 0.000000000
px 0 0.499921951 0.499921951 0.999843903 0.000000000
py 0 0.499921951 0.499921951 0.999843903 0.000000000
pz 0 0.499921953 0.499921953 0.999843906 0.000000000
sum over m 1.499765855 1.499765855 2.999531711 0.000000000
px 1 0.000078048 0.000078048 0.000156096 0.000000000
py 1 0.000078048 0.000078048 0.000156096 0.000000000
pz 1 0.000078048 0.000078048 0.000156096 0.000000000
sum over m 0.000234145 0.000234145 0.000468289 0.000000000
sum over m+mul 1.500000000 1.500000000 3.000000000 0.000000000
d3z^2-r^2 0 0.000000000 0.000000000 0.000000000 0.000000000
dx^2-y^2 0 0.000000000 0.000000000 0.000000000 0.000000000
dxy 0 0.000000000 0.000000000 0.000000000 0.000000000
dxz 0 0.000000000 0.000000000 0.000000000 0.000000000
dyz 0 0.000000000 0.000000000 0.000000000 0.000000000
sum over m 0.000000000 0.000000000 0.000000000 0.000000000
sum over m+mul 0.000000000 0.000000000 0.000000000 0.000000000我们计算磷原子的电荷变化
1.888594771+3.051110471+0.412470444-5=0.3522e
电荷转移的另一种计算方法
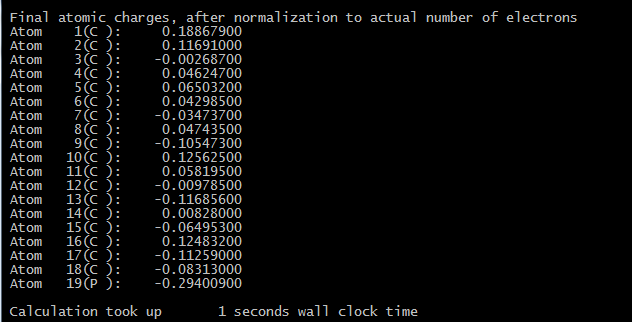
在把算好的文件.tden.cube为后缀的文件,打好包传到windows系统里,用multiwfn3.7来打开它(3.8版本就用不了VDD直接打开cube文件了),计算Voronoi deformation density (VDD) atom population,得到的结果是磷原子是施主:
Multiwfn3.7下载:
链接:https://pan.baidu.com/s/1C58vIVDM3fA5VckCoLifoA?pwd=1k9c
提取码:1k9c
–来自百度网盘超级会员V6的分享
气体分子HF和石墨烯之间具有很小的吸附能级很大的距离,这显示它们之间的吸附是很弱的。然而,在用Ti原子修饰石墨烯之后,吸附变强很多。对HF和H2S的吸附减小了twin石墨烯的能量。通过研究发现,将Ti嵌入twin石墨烯增长了石墨烯的吸附能力。所以可以考虑钛掺杂的石墨烯来作为这些有毒气体的探测。
近年来,固态气体传感器对有毒气体的探测具有高敏感度、低成本和小型化的优势,所以受到重点关注。
新材料的发现为气体探测提供了新的发展方向。以石墨烯为代表的碳基纳米材料在室温下对有毒气体很敏感。石墨烯基的气体探测器可以探测很多气体例如CO,CO2,NO,NO2,HF,H2S,H2CO,NH3等等。传感机制基于被吸附分子的电荷转移,这些电荷在石墨烯表面起着施主或者受主的作用。最近的研究表明掺杂可以增强石墨烯的探测敏感性。一些掺杂原子B,N,Al,Si等都已经被研究过了。
最近,一种新的叫做twin石墨烯的二维碳烯被引出。可以通过声子散射谱和结合能的计算来验证这种结构的稳定性。完整声子散射谱没有虚部,确认了twin石墨烯的动态稳定性。
其他吸附N原子:
将磷原子替换为氮原子,所用仿真代码在Graphene_N_distance,写入Graphene_15.dat,得到的结果如下。
1.0 -113.863888022818
1.1 -113.881310056625
1.2 -113.892801137945
1.3 -113.896821417428
1.4 -113.896387280476
1.55 -113.884201339466
1.56 -113.888451131785
1.57 -113.888227811492
1.58 -113.897860787325
1.59 -113.896765174184
1.6 -113.894480141191
1.61 -113.886988390561
1.62 -113.890855781931
1.63 -113.896235473871
1.64 -113.889588798824
1.65 -113.887667849867
1.7 -113.891837438702
1.8 -113.870282735787
1.9 -113.865234799820
2.0 -113.850019923809
2.1 -113.850613364301
2.2 -113.827071862439
2.3 -113.819898051190
2.4 -113.800121497027
2.5 -113.816869657681
2.6 -113.830216614218
2.7 -113.801684529095
2.8 -113.799694107760
2.9 -113.818978406898
3.0 -113.817951698737
其他吸附_NH3:
除了气体,固体分子也可以吸附到石墨烯表面,详细参考中科院的一篇博士论文【1】。
接下来,我们可以继续用氨气(NH3)来继续进行吸附实验。
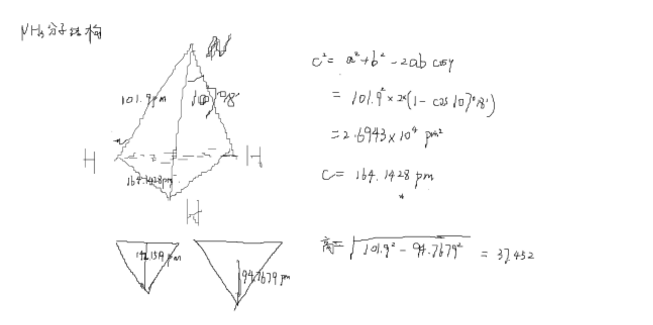
NH3的分子结构为三角锥形,夹角为107度18分,N-H键长为101.9pm。
#!bin/bash
if [ ! -f "energy.dat" ];then
echo 'energy.dat not exist'
else
rm -rf energy.dat
fi
for d in 2.6 2.7 2.8 2.9 3.0
do
typeset dd=$(echo "$d+0.3752"|bc)
cat << EOF > Graphene_NH3_0.dat
#
# File Name
#
System.CurrrentDirectory ./ # default=./
System.Name Graphene_NH3_0
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 1 # default=1 (0-2)
#
# Definition of Atomic Species
#
Species.Number 3
<Definition.of.Atomic.Species
H H5.0-s1 H_PBE13
C C6.0-s2p2d1 C_PBE13
N N7.0-s2p2d1 N_CA13
Definition.of.Atomic.Species>
#
# Atoms
#
Atoms.Number 22
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C 0.00000000 0.710000000 0.000000000 2.0 2.0
2 C 0.00000000 -0.71000000 0.000000000 2.0 2.0
3 C 1.22980000 1.420000000 0.000000000 2.0 2.0
4 C 1.22980000 -1.42000000 0.000000000 2.0 2.0
5 C -1.2298000 1.420000000 0.000000000 2.0 2.0
6 C -1.2298000 -1.42000000 0.000000000 2.0 2.0
7 C -2.4559120 0.71000000 0.000000000 2.0 2.0
8 C -2.4559120 -0.71000000 0.000000000 2.0 2.0
9 C -3.6893000 -1.42000000 0.000000000 2.0 2.0
10 C -3.6893000 -2.84000000 0.000000000 2.0 2.0
11 C -1.2298000 -2.84000000 0.000000000 2.0 2.0
12 C -4.9190000 -3.55000000 0.000000000 2.0 2.0
13 C -2.4595120 -3.55000000 0.000000000 2.0 2.0
14 C 0.00000000 -3.55000000 0.000000000 2.0 2.0
15 C 1.22980000 -2.84000000 0.000000000 2.0 2.0
16 C 2.45951200 0.710000000 0.000000000 2.0 2.0
17 C 2.45951200 -0.71000000 0.000000000 2.0 2.0
18 C 3.68930000 1.420000000 0.000000000 2.0 2.0
19 H 0.28230000 0.000000000 $d 0.5 0.5
20 H 1.70380000 0.820700000 $d 0.5 0.5
21 H 1.70380000 -0.82070000 $d 0.5 0.5
22 N 1.22980000 0.000000000 $dd 2.5 2.5
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors # unit=Ang.
4.91900000000000 -8.5200000000000 0.00000000000000
4.91900000000000 8.52000000000000 0.00000000000000
0.00000000000000 0.00000000000000 100.000000000000
Atoms.UnitVectors>
#
# SCF or Electronic System
#
scf.XcType GGA-PBE # LDA|LSDA-CA|LSDA-PW|GGA-PBE
scf.SpinPolarization Off # On|Off|NC
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 200.0 # default=150 (Ry)
scf.maxIter 60 # default=40
scf.EigenvalueSolver band # DC|GDC|Cluster|Band
scf.Kgrid 5 5 1 # means 4x4x4
scf.Mixing.Type rmm-diis # Simple|Rmm-Diis|Gr-Pulay|Kerker|Rmm-Diisk
scf.Init.Mixing.Weight 0.010 # default=0.30
scf.Min.Mixing.Weight 0.001 # default=0.001
scf.Max.Mixing.Weight 0.002 # default=0.40
scf.Mixing.History 7 # default=5
scf.Mixing.StartPulay 5 # default=6
scf.criterion 1.0e-9 # default=1.0e-6 (Hartree)
#
# MD or Geometry Optimization
#
MD.Type nomd # Nomd|Opt|NVE|NVT_VS|NVT_NH
MD.maxIter 1 # default=1
MD.TimeStep 0.5 # default=0.5 (fs)
MD.Opt.criterion 1.0e-4 # default=1.0e-4 (Hartree/bohr)
Band.dispersion on
Band.Nkpath 3
<Band.kpath
25 0.5 0.0 0.0 0.333 0.333 0.0 M K
25 0.333 0.333 0.0 0.0 0.0 0.0 K G
25 0.0 0.0 0.0 0.5 0.0 0.0 G M
Band.kpath>
#
# partial charge calculation
#
partial.charge on # on|off, default=off
partial.charge.energy.window 2.0 # in eV
#
# DOS and PDOS
#
Dos.fileout on # on|off, default=off
Dos.Erange -20.0 20.0 # default = -20 20
Dos.Kgrid 10 10 1 # default = Kgrid1 Kgrid2 Kgrid3
EOF
#if [ ! -f 'Graphene_NH3_0.out' ];then
./openmx Graphene_NH3_0.dat
#fi
#echo "d=$d " >> energy.dat
E=$(grep "Utot" Graphene_NH3_0.out|head -1| awk '{print $2}')
echo "$E " >> energy.dat
done在编制好了代码之后可以进行仿真了,距离——能量对比如下:
1.4 -115.653467139640
1.5 -115.701638908058
1.6 -115.739222247901
1.7 -115.764868660675
1.8 -115.790528960715
1.9 -115.806674400827
2.0 -115.818455984077
2.1 -115.827196317440
2.2 -115.828313924386
2.3 -115.837691236909
2.4 -115.840538996064
2.5 -115.843013234110
2.6 -115.843842480209
2.7 -115.845254359644
2.8 -115.834743395334
2.9 -115.837554660607
3.0 -115.844700972825
3.1 -115.845964684754
3.2 -115.845497336368
3.3 -115.845451641914
3.4 -115.845243558681
3.5 -115.844136790282电子电荷转移,我们粘贴出在NH3和石墨烯距离2.7埃的时候,三个氢原子和一个氮原子的电荷
19 H Up spin Down spin Sum Diff
multiple
s 0 0.452660617 0.452660617 0.905321234 0.000000000
sum over m 0.452660617 0.452660617 0.905321234 0.000000000
sum over m+mul 0.452660617 0.452660617 0.905321234 0.000000000
20 H Up spin Down spin Sum Diff
multiple
s 0 0.450287070 0.450287070 0.900574139 0.000000000
sum over m 0.450287070 0.450287070 0.900574139 0.000000000
sum over m+mul 0.450287070 0.450287070 0.900574139 0.000000000
21 H Up spin Down spin Sum Diff
multiple
s 0 0.449876901 0.449876901 0.899753801 0.000000000
sum over m 0.449876901 0.449876901 0.899753801 0.000000000
sum over m+mul 0.449876901 0.449876901 0.899753801 0.000000000
22 N Up spin Down spin Sum Diff
multiple
s 0 0.676576802 0.676576802 1.353153604 0.000000000
sum over m 0.676576802 0.676576802 1.353153604 0.000000000
s 1 -0.013567510 -0.013567510 -0.027135020 0.000000000
sum over m -0.013567510 -0.013567510 -0.027135020 0.000000000
sum over m+mul 0.663009292 0.663009292 1.326018584 0.000000000
px 0 0.555744068 0.555744068 1.111488135 0.000000000
py 0 0.557993782 0.557993782 1.115987565 0.000000000
pz 0 0.879635113 0.879635113 1.759270226 0.000000000
sum over m 1.993372963 1.993372963 3.986745926 0.000000000
px 1 -0.015638240 -0.015638240 -0.031276480 0.000000000
py 1 -0.015583137 -0.015583137 -0.031166274 0.000000000
pz 1 0.007163419 0.007163419 0.014326837 0.000000000
sum over m -0.024057959 -0.024057959 -0.048115917 0.000000000
sum over m+mul 1.969315004 1.969315004 3.938630009 0.000000000
d3z^2-r^2 0 0.001929680 0.001929680 0.003859360 0.000000000
dx^2-y^2 0 0.002440975 0.002440975 0.004881951 0.000000000
dxy 0 0.002414744 0.002414744 0.004829488 0.000000000
dxz 0 0.007893864 0.007893864 0.015787728 0.000000000
dyz 0 0.007903219 0.007903219 0.015806437 0.000000000
sum over m 0.022582482 0.022582482 0.045164964 0.000000000
sum over m+mul 0.022582482 0.022582482 0.045164964 0.000000000计算过程:
3H+1N :0.905321234+0.900574139+0.899753801+1.326018584+3.938630009+0.045164964-3-5
= 0.0155
这个结果表明NH3反而吸收了石墨烯表面的电子,与我们的预期不符,要予以改进。接下来我们实验图6后面一组数据,命名为Graphene_NH3_1_distance.sh:
#!bin/bash
if [ ! -f "energy.dat" ];then
echo 'energy.dat not exist'
else
rm -rf energy.dat
fi
for d in 1.0 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 2.0 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 2.9 3.0 3.1 3.2 3.3 3.4 3.5
do
typeset dd=$(echo "$d+0.3752"|bc)
cat << EOF > Graphene_NH3_1.dat
#
# File Name
#
System.CurrrentDirectory ./ # default=./
System.Name Graphene_NH3_1
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 1 # default=1 (0-2)
#
# Definition of Atomic Species
#
Species.Number 3
<Definition.of.Atomic.Species
H H5.0-s1 H_PBE13
C C6.0-s2p2d1 C_PBE13
N N7.0-s2p2d1 N_CA13
Definition.of.Atomic.Species>
#
# Atoms
#
Atoms.Number 22
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C 0.00000000 0.710000000 0.000000000 2.0 2.0
2 C 0.00000000 -0.71000000 0.000000000 2.0 2.0
3 C 1.22980000 1.420000000 0.000000000 2.0 2.0
4 C 1.22980000 -1.42000000 0.000000000 2.0 2.0
5 C -1.2298000 1.420000000 0.000000000 2.0 2.0
6 C -1.2298000 -1.42000000 0.000000000 2.0 2.0
7 C -2.4559120 0.71000000 0.000000000 2.0 2.0
8 C -2.4559120 -0.71000000 0.000000000 2.0 2.0
9 C -3.6893000 -1.42000000 0.000000000 2.0 2.0
10 C -3.6893000 -2.84000000 0.000000000 2.0 2.0
11 C -1.2298000 -2.84000000 0.000000000 2.0 2.0
12 C -4.9190000 -3.55000000 0.000000000 2.0 2.0
13 C -2.4595120 -3.55000000 0.000000000 2.0 2.0
14 C 0.00000000 -3.55000000 0.000000000 2.0 2.0
15 C 1.22980000 -2.84000000 0.000000000 2.0 2.0
16 C 2.45951200 0.710000000 0.000000000 2.0 2.0
17 C 2.45951200 -0.71000000 0.000000000 2.0 2.0
18 C 3.68930000 1.420000000 0.000000000 2.0 2.0
19 H 0.40910000 0.473800000 $d 0.5 0.5
20 H 2.05050000 0.473800000 $d 0.5 0.5
21 H 1.22980000 -0.94767900 $d 0.5 0.5
22 N 1.22980000 0.000000000 $dd 2.5 2.5
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors # unit=Ang.
4.91900000000000 -8.5200000000000 0.00000000000000
4.91900000000000 8.52000000000000 0.00000000000000
0.00000000000000 0.00000000000000 100.000000000000
Atoms.UnitVectors>
#
# SCF or Electronic System
#
scf.XcType GGA-PBE # LDA|LSDA-CA|LSDA-PW|GGA-PBE
scf.SpinPolarization Off # On|Off|NC
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 200.0 # default=150 (Ry)
scf.maxIter 60 # default=40
scf.EigenvalueSolver band # DC|GDC|Cluster|Band
scf.Kgrid 5 5 1 # means 4x4x4
scf.Mixing.Type rmm-diis # Simple|Rmm-Diis|Gr-Pulay|Kerker|Rmm-Diisk
scf.Init.Mixing.Weight 0.010 # default=0.30
scf.Min.Mixing.Weight 0.001 # default=0.001
scf.Max.Mixing.Weight 0.002 # default=0.40
scf.Mixing.History 7 # default=5
scf.Mixing.StartPulay 5 # default=6
scf.criterion 1.0e-9 # default=1.0e-6 (Hartree)
#
# MD or Geometry Optimization
#
MD.Type nomd # Nomd|Opt|NVE|NVT_VS|NVT_NH
MD.maxIter 1 # default=1
MD.TimeStep 0.5 # default=0.5 (fs)
MD.Opt.criterion 1.0e-4 # default=1.0e-4 (Hartree/bohr)
Band.dispersion on
Band.Nkpath 3
<Band.kpath
25 0.5 0.0 0.0 0.333 0.333 0.0 M K
25 0.333 0.333 0.0 0.0 0.0 0.0 K G
25 0.0 0.0 0.0 0.5 0.0 0.0 G M
Band.kpath>
#
# partial charge calculation
#
partial.charge on # on|off, default=off
partial.charge.energy.window 2.0 # in eV
#
# DOS and PDOS
#
Dos.fileout on # on|off, default=off
Dos.Erange -20.0 20.0 # default = -20 20
Dos.Kgrid 10 10 1 # default = Kgrid1 Kgrid2 Kgrid3
EOF
#if [ ! -f 'Graphene_NH3_1.out' ];then
./openmx Graphene_NH3_1.dat
#fi
#echo "d=$d " >> energy.dat
E=$(grep "Utot" Graphene_NH3_1.out|head -1| awk '{print $2}')
echo "$E " >> energy.dat
done计算结果如下:
1.0 -115.256426780835
1.1 -115.385714029143
1.2 -115.492372981170
1.3 -115.578811993670
1.4 -115.642551688750
1.5 -115.694639225808
1.6 -115.735622929256
1.7 -115.764632606099
1.8 -115.787523550056
1.9 -115.804429430505
2.0 -115.817029344927
2.1 -115.824243974204
2.2 -115.832738446847
2.3 -115.835606825665
2.4 -115.840480173090
2.5 -115.842904536583
2.6 -115.827758802881
2.7 -115.831914927275
2.8 -115.831208763502
2.9 -115.844459025825
3.0 -115.845807970795
3.1 -115.844586744097
3.2 -115.845640108537
3.3 -115.837813019187
3.4 -115.844603798909
3.5 -115.845182291746将NH3分子放到一个碳原子正上方,而不是放在苯环的中心试试:
除了文件名改成了Graphene_NH3_2_distance.sh还有文件内部的文件名定义,只修改NH3的原子位置那四行就可以了:
19 H 0.40910000 -0.94620000 $d 0.5 0.5
20 H 2.05050000 -0.94620000 $d 0.5 0.5
21 H 1.22980000 -2.36770000 $d 0.5 0.5
22 N 1.22980000 -1.42000000 $dd 2.5 2.5新的仿真结果:
1.0 -115.388039744742
1.1 -115.490890692747
1.2 -115.554995484459
1.3 -115.611496656116
1.4 -115.651190890522
1.5 -115.698167582228
1.6 -115.729692679203
1.7 -115.756474342970
1.8 -115.781225546577
1.9 -115.797342197904
2.0 -115.811012794682
2.1 -115.818195242985
2.2 -115.826908014851
2.3 -115.833167603346
2.4 -115.821003115501
2.5 -115.839993655785
2.6 -115.837559799331
2.7 -115.844701463077
2.8 -115.845304727418
2.9 -115.829814098382
3.0 -115.842089849298
3.1 -115.835773933784
3.2 -115.844721199815
3.3 -115.836208912303
3.4 -115.838345843247
3.5 -115.832028101333接下来,进行最后的H下N上的设置,也就是说,N原子放在C原子正上方,H原子指向苯环中心。
19 H -0.8207000 -0.23620000 $d 0.5 0.5
20 H 0.82070000 -0.23620000 $d 0.5 0.5
21 H 0.00000000 -1.65770000 $d 0.5 0.5
22 N 0.00000000 -0.71000000 $dd 2.5 2.5得到的结果如下:
1.0 -115.428664500354
1.1 -115.511038066138
1.2 -115.576078509646
1.3 -115.628478121016
1.4 -115.675305123328
1.5 -115.712793032016
1.6 -115.743883638842
1.7 -115.766336348537
1.8 -115.787888938649
1.9 -115.801791060515
2.0 -115.809560184875
2.1 -115.818910142569
2.2 -115.826530775014
2.3 -115.835107087414
2.4 -115.838194954559
2.5 -115.832267586663
2.6 -115.835676324103
2.7 -115.843345308068
2.8 -115.842332536415
2.9 -115.840080726347
3.0 -115.835712300328
3.1 -115.842712131661
3.2 -115.840273816466
3.3 -115.841709166625
3.4 -115.836936554149
3.5 -115.833885207559我们将上面的四个文件的结果画在一起:
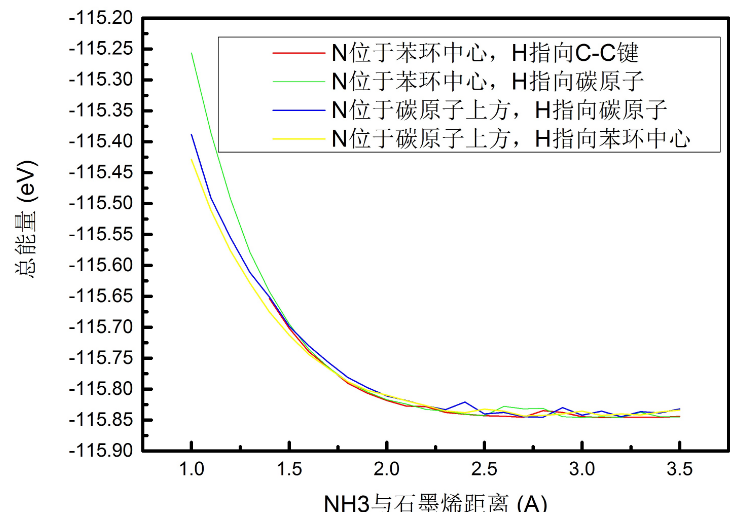
将NH3的方向倒转,可以得到更加全面的设置图。
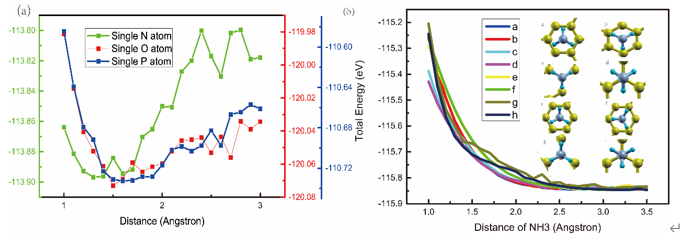
Distance of NH3 a b c d e f g h
1 — -115.256 -115.388 -115.429 -115.29 -115.264 -115.205 -115.245
1.1 — -115.386 -115.491 -115.511 -115.378 -115.361 -115.407 -115.444
1.2 — -115.492 -115.555 -115.576 -115.457 -115.454 -115.537 -115.565
1.3 — -115.579 -115.611 -115.628 -115.539 -115.535 -115.599 -115.64
1.4 -115.653 -115.643 -115.651 -115.675 -115.603 -115.601 -115.66 -115.681
1.5 -115.702 -115.695 -115.698 -115.713 -115.656 -115.655 -115.667 -115.71
1.6 -115.739 -115.736 -115.73 -115.744 -115.698 -115.697 -115.689 -115.728
1.7 -115.765 -115.765 -115.756 -115.766 -115.733 -115.732 -115.703 -115.74
1.8 -115.791 -115.788 -115.781 -115.788 -115.754 -115.757 -115.735 -115.756
1.9 -115.807 -115.804 -115.797 -115.802 -115.781 -115.781 -115.753 -115.764
2 -115.818 -115.817 -115.811 -115.81 -115.796 -115.796 -115.768 -115.776
2.1 -115.827 -115.824 -115.818 -115.819 -115.81 -115.811 -115.781 -115.799
2.2 -115.828 -115.833 -115.827 -115.827 -115.82 -115.82 -115.803 -115.807
2.3 -115.838 -115.836 -115.833 -115.835 -115.827 -115.826 -115.787 -115.819
2.4 -115.841 -115.84 -115.821 -115.838 -115.833 -115.833 -115.82 -115.826
2.5 -115.843 -115.843 -115.84 -115.832 -115.834 -115.837 -115.817 -115.831
2.6 -115.844 -115.828 -115.838 -115.836 -115.841 -115.83 -115.832 -115.837
2.7 -115.845 -115.832 -115.845 -115.843 -115.842 -115.831 -115.833 -115.84
2.8 -115.835 -115.831 -115.845 -115.842 -115.842 -115.839 -115.829 -115.843
2.9 -115.838 -115.844 -115.83 -115.84 -115.845 -115.845 -115.825 -115.844
3 -115.845 -115.846 -115.842 -115.836 -115.846 -115.846 -115.838 -115.845
3.1 -115.846 -115.845 -115.836 -115.843 -115.842 -115.842 -115.841 -115.846
3.2 -115.845 -115.846 -115.845 -115.84 -115.832 -115.841 -115.837 -115.846
3.3 -115.845 -115.838 -115.836 -115.842 -115.829 -115.837 -115.836 -115.846
3.4 -115.845 -115.845 -115.838 -115.837 -115.842 -115.838 -115.834 -115.842
3.5 -115.844 -115.845 -115.832 -115.834 -115.832 -115.846 -115.835 -115.847
当我们把NH3与石墨烯的距离增大到5埃的时候,总能量就在-115.843526996035eV左右,NH3的电荷转移计算值只有-4.5338e-05量级,可以忽略不计。所以从上面的图和计算可以得出,NH3在石墨烯表面吸附这整个系统就是一个能量从高到低,低到平的过程,并没有像那些共价键一样有一个能量回升的过程。
在共价键的形成中,能量先减后增加,这也印证了共价键的稳定性。而类似这种吸附的过程是相当不稳固的。
如果我们将NH3的分子翻一个个,N原子朝下,三个H原子朝上,重新进行仿真。具体操作上,就是在相应的文件Grpahene_NH3_distance.sh中将$d和$dd互换得到Graphene_NH3_verse_distance.sh。距离为3埃的时候,从得到的结果看,来计算电荷转移
0.895722617+0.894030643+0.893591796+1.313380094+3.958885696+0.044766221-3-5=3.7707e-04
得到的总能量Utot = -115.845844566343 eV
从上面的仿真结果可以看出,NH3分子与石墨烯表面并没有明显的电荷转移。我们将NH3分子和石墨烯的距离减小到2.0,再次进行仿真
0.887053543+0.883764133+0.881693467+1.352295807+3.876562810+0.070644073-3-5=-0.048
得到的总能量为Utot=-115.796439833180 eV
黑磷晶体在石墨烯表面的吸附
在经历了上面的磷原子和NH3在石墨烯的表面吸附的例子,我们再次将黑磷的晶体放到石墨烯表面,黑磷用了10个原子,得到了以下的输入文件:
#!bin/bash
if [ ! -f "energy.dat" ];then
echo 'energy.dat not exist'
else
rm -rf energy.dat
fi
for d in 1.0
do
typeset d1=$(echo "$d+0.394200015664101"|bc)
typeset d2=$(echo "$d+3.98579998433590"|bc)
typeset d3=$(echo "$d+2.58420001566410"|bc)
typeset d4=$(echo "$d+1.79579998433590"|bc)
typeset d5=$(echo "$d+0.394200015664101"|bc)
typeset d6=$(echo "$d+3.98579998433590"|bc)
typeset d7=$(echo "$d+2.58420001566410"|bc)
typeset d8=$(echo "$d+1.79579998433590"|bc)
typeset d9=$(echo "$d+4.77420001566410"|bc)
typeset d10=$(echo "$d+4.77420001566410"|bc)
cat << EOF > Graphene_BP_distance.dat
#
# File Name
#
System.CurrrentDirectory ./ # default=./
System.Name Graphene_BP_distance
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 1 # default=1 (0-2)
#
# Definition of Atomic Species
#
Species.Number 2
<Definition.of.Atomic.Species
C C6.0-s2p2d1 C_PBE13
P P8.0-s2p2d1 P_CA13
Definition.of.Atomic.Species>
#
# Atoms
#
Atoms.Number 28
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C 0.00000000 0.710000000 0.000000000 2.0 2.0
2 C 0.00000000 -0.71000000 0.000000000 2.0 2.0
3 C 1.22980000 1.420000000 0.000000000 2.0 2.0
4 C 1.22980000 -1.42000000 0.000000000 2.0 2.0
5 C -1.2298000 1.420000000 0.000000000 2.0 2.0
6 C -1.2298000 -1.42000000 0.000000000 2.0 2.0
7 C -2.4559120 0.71000000 0.000000000 2.0 2.0
8 C -2.4559120 -0.71000000 0.000000000 2.0 2.0
9 C -3.6893000 -1.42000000 0.000000000 2.0 2.0
10 C -3.6893000 -2.84000000 0.000000000 2.0 2.0
11 C -1.2298000 -2.84000000 0.000000000 2.0 2.0
12 C -4.9190000 -3.55000000 0.000000000 2.0 2.0
13 C -2.4595120 -3.55000000 0.000000000 2.0 2.0
14 C 0.00000000 -3.55000000 0.000000000 2.0 2.0
15 C 1.22980000 -2.84000000 0.000000000 2.0 2.0
16 C 2.45951200 0.710000000 0.000000000 2.0 2.0
17 C 2.45951200 -0.71000000 0.000000000 2.0 2.0
18 C 3.68930000 1.420000000 0.000000000 2.0 2.0
19 P 0.00000000000000 1.02899997308850 $d1 2.5 2.5
20 P 0.00000000000000 -1.0289999730885 $d2 2.5 2.5
21 P 1.65500000000000 -1.0289999730885 $d3 2.5 2.5
22 P 1.65500000000000 1.02899997308850 $d4 2.5 2.5
23 P 3.31000000000000 1.02899997308850 $d5 2.5 2.5
24 P 3.31000000000000 -1.0289999730885 $d6 2.5 2.5
25 P 4.96500000000000 -1.0289999730885 $d7 2.5 2.5
26 P 4.96500000000000 1.02899997308850 $d8 2.5 2.5
27 P 0.00000000000000 1.02899997308850 $d9 2.5 2.5
28 P 3.31000000000000 1.02899997308850 $d10 2.5 2.5
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors # unit=Ang.
4.91900000000000 -8.5200000000000 0.00000000000000
4.91900000000000 8.52000000000000 0.00000000000000
0.00000000000000 0.00000000000000 100.000000000000
Atoms.UnitVectors>
#
# SCF or Electronic System
#
scf.XcType GGA-PBE # LDA|LSDA-CA|LSDA-PW|GGA-PBE
scf.SpinPolarization Off # On|Off|NC
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 200.0 # default=150 (Ry)
scf.maxIter 60 # default=40
scf.EigenvalueSolver band # DC|GDC|Cluster|Band
scf.Kgrid 5 5 1 # means 4x4x4
scf.Mixing.Type rmm-diis # Simple|Rmm-Diis|Gr-Pulay|Kerker|Rmm-Diisk
scf.Init.Mixing.Weight 0.010 # default=0.30
scf.Min.Mixing.Weight 0.001 # default=0.001
scf.Max.Mixing.Weight 0.002 # default=0.40
scf.Mixing.History 7 # default=5
scf.Mixing.StartPulay 5 # default=6
scf.criterion 1.0e-9 # default=1.0e-6 (Hartree)
#
# MD or Geometry Optimization
#
MD.Type nomd # Nomd|Opt|NVE|NVT_VS|NVT_NH
MD.maxIter 1 # default=1
MD.TimeStep 0.5 # default=0.5 (fs)
MD.Opt.criterion 1.0e-4 # default=1.0e-4 (Hartree/bohr)
Band.dispersion on
Band.Nkpath 3
<Band.kpath
25 0.5 0.0 0.0 0.333 0.333 0.0 M K
25 0.333 0.333 0.0 0.0 0.0 0.0 K G
25 0.0 0.0 0.0 0.5 0.0 0.0 G M
Band.kpath>
#
# partial charge calculation
#
partial.charge on # on|off, default=off
partial.charge.energy.window 2.0 # in eV
#
# DOS and PDOS
#
Dos.fileout on # on|off, default=off
Dos.Erange -20.0 20.0 # default = -20 20
Dos.Kgrid 10 10 1 # default = Kgrid1 Kgrid2 Kgrid3
EOF
#if [ ! -f 'Graphene_13.out' ];then
./openmx Graphene_BP_distance.dat
#fi
#echo "d=$d " >> energy.dat
E=$(grep "Utot" Graphene_BP_distance.out|head -1| awk '{print $2}')
echo "$E " >> energy.dat
done计算这个文件,我们通过统计10个P原子的核外电子,发现10个磷原子确实是电子受体,吸收了0.7502个电子。回忆NH3的结果,NH3在距离石墨烯很近的时候确实是施主。这个结果就和实验相符合了。不用算也可以知道,1埃的距离是近了,能量还并不是最低的。
接下来,我想写一个脚本,来自动统计.out文件中的电荷转移。
#!bin/bash
if [ ! -f "energy.dat" ];then
echo 'energy.dat not exist'
else
rm -rf energy.dat
fi
for d in 1.5
do
typeset d1=$(echo "$d+2.05799994617701"|bc)
typeset d2=$(echo "$d+3.98579998433590"|bc)
typeset d3=$(echo "$d+2.58420001566410"|bc)
typeset d4=$(echo "$d+1.79579998433590"|bc)
typeset d5=$(echo "$d+0.394200015664101"|bc)
typeset d6=$(echo "$d+3.98579998433590"|bc)
typeset d7=$(echo "$d+2.58420001566410"|bc)
typeset d8=$(echo "$d+1.79579998433590"|bc)
typeset d9=$(echo "$d+4.77420001566410"|bc)
typeset d10=$(echo "$d+4.77420001566410"|bc)
cat << EOF > Graphene_BP_distance.dat
#
# File Name
#
System.CurrrentDirectory ./ # default=./
System.Name Graphene_BP_distance
level.of.stdout 1 # default=1 (1-3)
level.of.fileout 1 # default=1 (0-2)
#
# Definition of Atomic Species
#
Species.Number 2
<Definition.of.Atomic.Species
C C6.0-s2p2d1 C_PBE13
P P8.0-s2p2d1 P_CA13
Definition.of.Atomic.Species>
#
# Atoms
#
Atoms.Number 28
Atoms.SpeciesAndCoordinates.Unit Ang # Ang|AU
<Atoms.SpeciesAndCoordinates # Unit=Ang.
1 C 0.00000000 0.710000000 0.000000000 2.0 2.0
2 C 0.00000000 -0.71000000 0.000000000 2.0 2.0
3 C 1.22980000 1.420000000 0.000000000 2.0 2.0
4 C 1.22980000 -1.42000000 0.000000000 2.0 2.0
5 C -1.2298000 1.420000000 0.000000000 2.0 2.0
6 C -1.2298000 -1.42000000 0.000000000 2.0 2.0
7 C -2.4559120 0.71000000 0.000000000 2.0 2.0
8 C -2.4559120 -0.71000000 0.000000000 2.0 2.0
9 C -3.6893000 -1.42000000 0.000000000 2.0 2.0
10 C -3.6893000 -2.84000000 0.000000000 2.0 2.0
11 C -1.2298000 -2.84000000 0.000000000 2.0 2.0
12 C -4.9190000 -3.55000000 0.000000000 2.0 2.0
13 C -2.4595120 -3.55000000 0.000000000 2.0 2.0
14 C 0.00000000 -3.55000000 0.000000000 2.0 2.0
15 C 1.22980000 -2.84000000 0.000000000 2.0 2.0
16 C 2.45951200 0.710000000 0.000000000 2.0 2.0
17 C 2.45951200 -0.71000000 0.000000000 2.0 2.0
18 C 3.68930000 1.420000000 0.000000000 2.0 2.0
19 P 0.394200015664101 0 $d1 2.5 2.5
20 P 3.98579998433590 0 $d 2.5 2.5
21 P 2.58420001566410 1.65500000000000 $d 2.5 2.5
22 P 1.79579998433590 1.65500000000000 $d1 2.5 2.5
23 P 0.394200015664101 3.31000000000000 $d1 2.5 2.5
24 P 3.98579998433590 3.31000000000000 $d 2.5 2.5
25 P 2.58420001566410 4.96500000000000 $d 2.5 2.5
26 P 1.79579998433590 4.96500000000000 $d1 2.5 2.5
27 P 4.77420001566410 0 $d1 2.5 2.5
28 P 4.77420001566410 3.31000000000000 $d1 2.5 2.5
Atoms.SpeciesAndCoordinates>
Atoms.UnitVectors.Unit Ang # Ang|AU
<Atoms.UnitVectors # unit=Ang.
4.91900000000000 -8.5200000000000 0.00000000000000
4.91900000000000 8.52000000000000 0.00000000000000
0.00000000000000 0.00000000000000 100.000000000000
Atoms.UnitVectors>
#
# SCF or Electronic System
#
scf.XcType GGA-PBE # LDA|LSDA-CA|LSDA-PW|GGA-PBE
scf.SpinPolarization Off # On|Off|NC
scf.ElectronicTemperature 300.0 # default=300 (K)
scf.energycutoff 200.0 # default=150 (Ry)
scf.maxIter 60 # default=40
scf.EigenvalueSolver band # DC|GDC|Cluster|Band
scf.Kgrid 5 5 1 # means 4x4x4
scf.Mixing.Type rmm-diis # Simple|Rmm-Diis|Gr-Pulay|Kerker|Rmm-Diisk
scf.Init.Mixing.Weight 0.010 # default=0.30
scf.Min.Mixing.Weight 0.001 # default=0.001
scf.Max.Mixing.Weight 0.002 # default=0.40
scf.Mixing.History 7 # default=5
scf.Mixing.StartPulay 5 # default=6
scf.criterion 1.0e-9 # default=1.0e-6 (Hartree)
#
# MD or Geometry Optimization
#
MD.Type nomd # Nomd|Opt|NVE|NVT_VS|NVT_NH
MD.maxIter 1 # default=1
MD.TimeStep 0.5 # default=0.5 (fs)
MD.Opt.criterion 1.0e-4 # default=1.0e-4 (Hartree/bohr)
Band.dispersion on
Band.Nkpath 3
<Band.kpath
25 0.5 0.0 0.0 0.333 0.333 0.0 M K
25 0.333 0.333 0.0 0.0 0.0 0.0 K G
25 0.0 0.0 0.0 0.5 0.0 0.0 G M
Band.kpath>
#
# partial charge calculation
#
partial.charge on # on|off, default=off
partial.charge.energy.window 2.0 # in eV
#
# DOS and PDOS
#
Dos.fileout on # on|off, default=off
Dos.Erange -20.0 20.0 # default = -20 20
Dos.Kgrid 10 10 1 # default = Kgrid1 Kgrid2 Kgrid3
EOF
#if [ ! -f 'Graphene_13.out' ];then
./openmx Graphene_BP_distance.dat
#fi
#echo "d=$d " >> energy.dat
E=$(grep "Utot" Graphene_BP_distance.out|head -1| awk '{print $2}')
typeset el=0
eval $(awk -F" " '/m\+mul/{if (NR>=3014) {ele+=$6}} END {printf"el=%2.4f",ele;}' Graphene_BP_distance.out)
echo "$d $E $el" >> energy.dat
done这里的$d2~d10没有用到,主要在
eval $(awk -F" " '/m\+mul/{if (NR>=3014) {ele+=$6}} END {printf"el=%2.4f",ele;}' Graphene_BP_distance.out)这一句,在3014行之后才都是P原子,所以要进行这个判断。如果不加这个判断,那么总电子数,就是P原子加上C原子的价电子,总共122个。关于eval和awk命令可以看这里:
linux常用命令
我们计算石墨烯上黑磷量子点的吸附,结果距离、能量、电子。文件中是10个P原子,总共有50个价电子。总结如下
1.0 -171.444528393218 52.1330
1.1 -171.930204236779 51.6530
1.2 -172.245896059095 51.5808
1.3 -172.495608125017 51.3968
1.4 -172.442344814281 51.1738
1.5 -172.442165645930 50.7591
1.7 -172.298518415490 50.7277
2.0 -172.066466368335 50.3074
2.5 -172.005642164082 50.2365
3.0 -172.200267039593 50.1833
3.1 -172.203625647829 50.0774
3.2 -172.268446553834 50.0930
3.3 -167.586555491771 49.8886
3.4 -171.919741464621 50.0851
3.5 -168.589033975806 50.0699
3.6 -167.488968595546 51.8452
3.7 -167.901320870206 50.0166
3.8 -168.164834494739 50.0730
3.9 -168.923881587129 49.9742
4.0 -166.148638362152 52.4947
4.5 -167.551750269218 49.8472
5.0 -167.772249597457 49.8495论文结构:
图:
1.无吸附,单石墨烯能带,态密度,分子云。
2. P单原子吸附能带,态密度,分子云。
3.P原子吸附的带隙——距离,吸附位置。
4.分波态密度。
5.变换分子NH3分子。角度,位置。
6.不同分子的电负性和实验对比。
7.电荷转移情况。
8.NEGF示意图,透射谱
9.NEGF IV,不同原子。
第二种思路
1.原胞设置,坐标系统
2.驰预距离,点位选择
3.能带,石墨烯,大胞,磷
4.大胞,PDOS,磷,PDOS
5.表格,能量,电荷转移
6.NEGF示意图
7.NEGF透射谱
8.NEGF IV 不同原子
六图一表版:
1.石墨烯能带,态密度,电子云 (问题:电子云指代不明)
2.晶格设置,原胞说明,距离驰预
3.P原子吸附的分波态密度,电子云,积分态密度(问题:分波态密度主题不突出)
4.BPQD在石墨烯上的转移曲线和It曲线(补上NH3的实验,调整和图5的位置)
5.NH3不同点位吸附图能量,BPQD和P原子吸附对比图
表:不同原子分子转移电荷数目,能量差
6.NEGF传输IV曲线,吸附前后影响
参考文献:
1.碳纳米材料的第一性原理研究